Setting parameters and run an experiment in software for numerical simulation of the crystal growth.
The "Run" panel of LeoMonteCrystal is to set technical parameters of numerical simulation experiment.
The Fig. 1 shows screenshot of the “Run” panel.
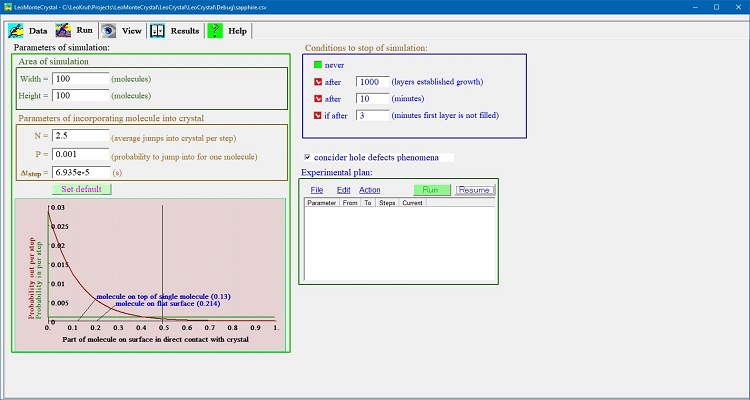
Fig. 1. Run panel contains groups of controls combined by functional
attributes for editing or displaying technical parameters of
numerical experiment.
"Area of simulation"
controls group permits defining dimensions of a matrix that represented area of surface numerical simulation is performed in.
|
|
||
| Fig. 2. Group of controls for setting size of matrix to perform simulation experiment. |

Selection of the size of simulation field is a matter of compromise as it is
almost always true when discussing numerical simulation. The larger field the
closer to reality (not always if we talking about nano size objects)
the smaller the faster simulation.
Usually one can presume that for most cases 100×100 and
large field is quite sufficient and 10×10 is too small.
Normally filed size should be large enough to accommodate at
least one critical nucleus. It means that at very smaller supercooling
when theoretical value of critical nuclei is approaching infinity
simulation field size should be as large as possible for stable
outcomes that putting hardware performance limitations on the method.
Time step and probability chart
|
|
||
| Fig. 3. Group of controls for setting time step at numerical simulation experiment and displaying probabilities for one molecule to be incorporated into crystal or jump out from it. Red line represents probability for molecule at surface to be emitting - locally stepping one layer back, green line gives probability to incorporate one molecule in crystal at the position making step ahead. |
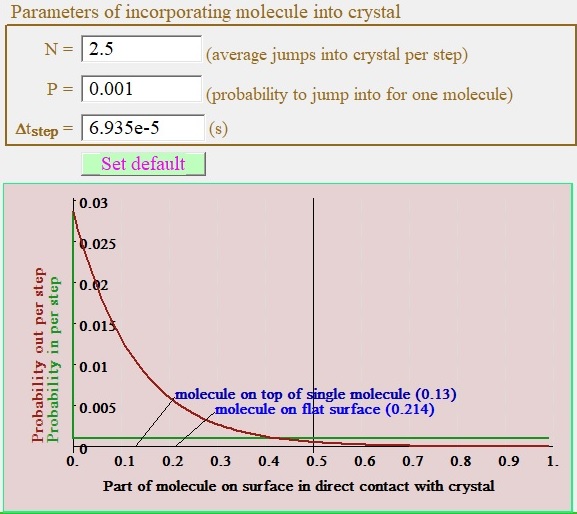
The group of controls for defining time steps value during numerical
simulation contains three hard interconnected parameters each
of them defined two others. User can edit any of them hit enter
key and other two will be update automatically.
The N value gives average number of molecules that will be
randomly jump into crystalline phase from outside to be incorporated at next
time step randomly at positions of whole simulation field.
Probability P for any specific molecule position to accept
molecule and move one layer ahead will be N divided on
number positions in the simulation field.
The value of the time step in seconds is probability
for a molecule to jump scaled by frequency coefficient calculated from model of
thermally activated reaction:
Δtstep = P / (γo * exp(-Ea/kT))
where γo - frequency of thermal vibrations of molecules in crystal and surrounded liquid phase, Ea - activation energy for jump incorporating into crystalline phase, k - Boltzmann's constant, T - temperature.
Pushing a button: "Set default" user asking a program to set parameters described above to the recommended values.
Most useful parameter to set experiment is to the probability P. Usually the value of probability 0.001 is the best fitting compromise between reliability of calculation and computation time necessary to reach stable results.
Depend on neighborhood surrounding of the molecule on crystal surface specifically value of part of molecule that is in direct contact with others near neighbors in crystal there are different probabilities to incorporate next molecule into crystal at this position or emitting one molecule into surrounding phase.
We use state of art proprietary algorithm for calculation of these probabilities of stepping ahead or back or staying unmoved as third alternative these are functions of on part of molecule surface in direct contact with other molecules of the crystal.
It's worth to note that probability to jump in - P from time step group is equal to probability to jump in in actual simulation only for the case when probability jump back is approaching zero - that is the situation of the "valley" like position on the crystal surface when part of molecule in direct contact with crystal approaching 1.
Depend on curve behavior of Pin(ΔSm) and Pout(ΔSm) curves on the Fig. 3 user can make education suggestion about adequacy of selecting value of P. Specifically if to chose value P too large like >0.1 one can see that at even at areas of values of ΔSm around 0.5 probability jump out could approach to 1 that could be clear sign of results distortion. Other way around the setting too small value of P<1e-5 could produce artifact effects connected with size of increment step in random number generator used in the program.
The chart of probabilities is the most powerful tool for user to exam correctness of setting parameters of numerical simulation.
Condition to stop simulation
controls group allow to set time oor distance limits for one numerical experiment:
|
|
||
| Fig. 4. Group of controls to set stop conditions for one experiment. |

User have options to let simulation run indefinitely long until computer permits or limit one experiment by distance of advance of crystal surface or by physical time of experiment or both whatever comes first. The option is mostly valuable when series of experiments are running automatically that could be essential for situations: a) large supercooling when experiment can be accomplished in very short physical computer time and there is no need to continue longer then let say 100 layers of growth; b) at small supercooling filling even several layers can take days so limiting time for one of experiment in whole series looks like reasonable.
User also have an option to include in the algorithm possibility of creation hole like defects when well like molecule structure is clogged at neck by other molecule creating empty space inside crystal.
Experimental plan control
displayed at Fig. 5:
|
|
||
| Fig. 5. Experimental plan control. |
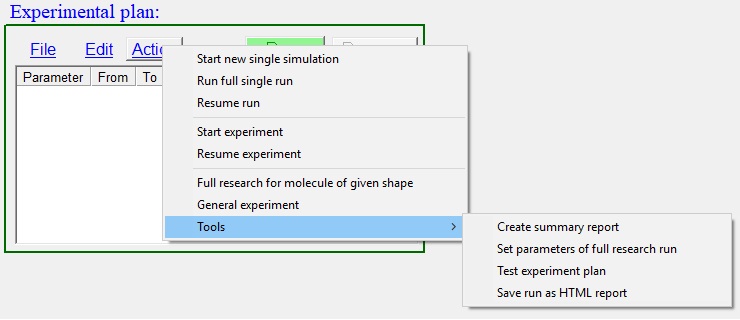
Group of controls are combined together permit: to run single numerical experiment or series of experiments follow users instructions.
A button "Run" starts a single experiment with currently set parameters. The experiment will start from smooth surface.
Pressing button "Pause/Resume" permits to pause a simulation at any point and then resume it with or without changing of conditions for simulation experiment. User can use it by variety reasons like for instance to learn how changing temperature affect crystal growth and estimating time for accommodation from one regime to another.
A menu and list of tasks at "Experiment plan" control can be use for setting series of numerical experiments to be run automatically. There is description of menu commands contained in the control:
-
File: - provides operations with file containing current
conditions of experiment plus information about loops of variables
defining experimental plan.
- New - creates blank file for saving later an experimental plan.
- Open - open file with experimental plan.
- Save - save current experimental plan including current point in its progress.
- Save As - save current experimental plan, including current point in execution of the variables loops, under new name.
- Delete - delete all loops in experimental plan.
-
Edit: - experimental plan contains loops of experiment
parameters to be executed in ascended order. Loops themselves
contains sub-loops defined with starting and ending point for
variable to try and number points in sub-loop. One variable can
have only one loop that itself can contain any reasonable number
of sub-loops.
- Add - by clicking on one of the following parameters user will add one sub-loop of corresponding parameter to the experimental plan. Depend on presence of the sub-loops of the variable default parameters of new sub-loop will be defined by program itself. User can change all parameters of sub-loop by clicking on the corresponding number and editing it. Under list of sub-loops there is visual representation of the points in the loops and current point is marked by the boxes.
-
General parameters:
- General supercooling - (unitless)
- General surface energy - (unitless)
-
Thermodynamics:
- Enthalpy (specific volume)
- Surface energy
- Temperature
- Concentration
-
Molecule:
- Volume
- Ratio a/b - (unitless)
- Ration b/c - (unitless)
- Delete - remove one highlighted sub-loop
-
Change order - move highlighted sub-loop to change order
of execution of nested loops. Position of the current point of
very highest loop is changed after execution of all points of
low order loop that will be tried again for new point. One can
see experimental plan as multidimensional (by number of loops)
matrix with uneven size of steps for one dimension (if there are more
than on sub-loops in it).
- Up
- Down
- Jump up to the top
Important to note that experiment points are in measurement units currently active at "Data" panel and will not be changed if user will modify used units. A recommended practice is set parameters in "Data" panel that will fit to one desirable experiment in series using convenient measurement units and then add one by one sub-loops of changeable in numerical experiments parameters.
Following are available parameters to range in loops:
-
Action:
- Start new single simulation - the same effect as pushing a button "Run"; one single simulation will start from smooth surface in neutral position.
- Start experiment - starts execution of experimental plan.
- Resume experiment - resumes the execution of the currently open experiment plan if it was stopped by user by clicking on the button "Pause" or if opened plan was interrupted before saving.
- Full research for molecule of given shape - automatically runs full research for molecule of given shape that is defined by ratios of molecule sizes in direction of its three axes. After clicking this item of "Action" menu program will produce full research for defined in Data panel shape of molecule varying general supercooling and general surface energy, two parameters are unequivocally defining roughness of the growing surface and mechanism of growth for the molecule of given shape. This command was used to gather a data collecting on the base of it approximation formulas for best presentation of surface roughness and coefficient reflecting influence of surface energy on theoretical growth rate by thermally activation surface reaction.
- General experiment - run a series of experiments aimed to cover all possible values of parameters to use result in finding approximation formulas. That was achieved by us already.
- Use one of the available tools: create summary of run; set parameters for full research run; dry test how works experimental plan; create report of one run as HTML page.